Das Apert-Syndrom ist eine seltene genetische Störung, die dazu führt, dass sich Gesichts- und Schädelknochen eines Fötus zu früh in ihrer Entwicklung verschmelzen.
Das Apert-Syndrom verursacht Gesichts- und Schädelanomalien, die zu Sehstörungen und Zahnproblemen führen können. Apert-Syndrom kann auch Anomalien in den Fingern und Zehen verursachen.
Dieser Artikel bietet einen Überblick über das Apert-Syndrom, einschließlich der Symptome, Behandlungen und Aussichten für diesen Zustand.
Was ist das Apert-Syndrom?
Apert-Syndrom ist eine Erkrankung, bei der die Schädelknochen zu früh verschmelzen, was sich auf die Form des Kopfes und des Gesichts auswirkt.
Menschen, die mit Apert-Syndrom geboren wurden, können aufgrund der abnormalen Form der Gesichts- und Schädelknochen Probleme mit ihrer Sehkraft und ihren Zähnen haben.
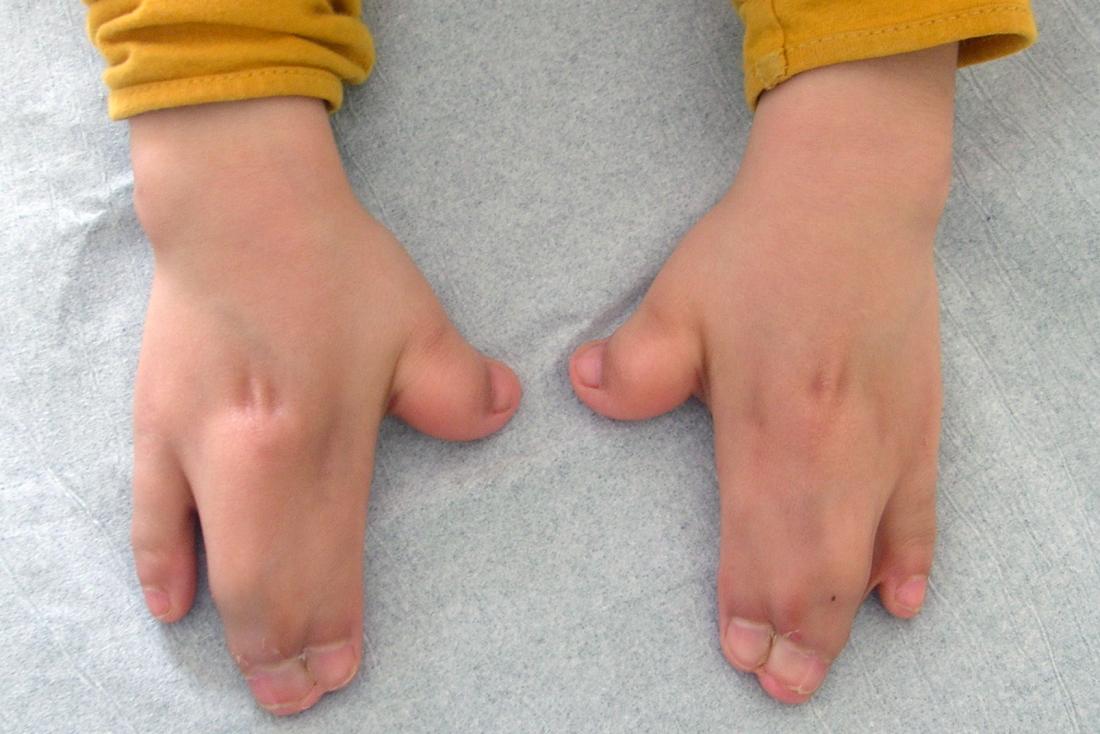
In vielen Fällen verschmelzen auch drei oder mehr Finger oder Zehen miteinander, was als Syndaktylie bezeichnet wird.
Apert-Syndrom ist eine genetische Störung. Es erscheint normalerweise ohne Familiengeschichte des Syndroms, aber es kann auch von einem Elternteil geerbt werden.
Symptome
Charakteristische Symptome des Apert-Syndroms sind:
- ein kegelförmiger Schädel, bekannt als Turribrachyzephalie
- ein Gesicht, das tief in der Mitte liegt
- Augen, die weit sind und sich nach außen wölben
- eine Schnabelnase
- ein unterentwickelter Oberkiefer, der dazu führen kann, dass die Zähne überfüllt werden
Die Gesichts- und Schädelanomalien können zu einigen Gesundheits- und Entwicklungsproblemen führen. Wenn nicht korrigiert, treten Sehprobleme oft als Folge von flachen Augenhöhlen auf. Der Oberkiefer ist in der Regel kleiner als der Durchschnitt, was zu Zahnproblemen führen kann, wenn die Zähne des Kindes wachsen.
Beim Apert-Syndrom ist der Schädel kleiner als gewöhnlich, was das sich entwickelnde Gehirn unter Druck setzen kann. Menschen mit Apert-Syndrom können einen durchschnittlichen Intellekt oder eine leichte bis mittelschwere intellektuelle Beeinträchtigung aufweisen.
Kinder, die mit dem Apert-Syndrom geboren wurden, haben oft Finger und Zehen mit Schwimmhäuten. Meistens sind drei Finger oder Zehen miteinander verschmolzen, aber manchmal kann eine ganze Reihe von Fingern oder Zehen mit Schwimmhäuten versehen sein. Seltener kann ein Kind zusätzliche Finger oder Zehen haben.
Zusätzliche Anzeichen und Symptome des Apert-Syndroms sind:
- Schwerhörigkeit
- schwere Akne
- starkes Schwitzen
- Fusion der Wirbelknochen im Nacken
- ölige Haut
- fehlende Haare in den Augenbrauen
- Wachstums- und Entwicklungsverzögerungen
- wiederkehrende Ohrinfektionen
- eine Gaumenspalte
- leichte bis mittelschwere geistige Behinderung
Ursachen

Apert-Syndrom ist eine Geburtsanomalie, die durch eine Mutation des Gens verursacht wird. Dies kann bei Babys ohne Familienanamnese auftreten, oder sie können sie von einem Elternteil erben.
Das Gen produziert ein Protein namens Fibroblasten-Wachstumsfaktor-Rezeptor 2. Dieses Protein hat viele wichtige Rollen in der Entwicklung eines Fötus, einschließlich einer Schlüsselrolle bei der Entwicklung von Knochenzellen.
Wenn diese Genmutation auftritt, signalisiert sie länger als üblich, was zu einer frühen Verschmelzung von Schädel, Gesicht, Füßen und Handknochen führt.
Mutationen am Gen können auch mehrere andere verwandte Erkrankungen verursachen, einschließlich:
- Pfeiffer-Syndrom
- Crouzon-Syndrom
- Jackson-Weiss-Syndrom
Häufigkeit und Vererbung
Apert-Syndrom ist eine extrem seltene Erkrankung. Die Anzahl der Personen, die sie haben, ist nicht bekannt, und die Schätzungen variieren zwischen den Quellen.
Die US-amerikanische National Library of Medicine schätzt, dass 1 von 65.000 bis 88.000 Neugeborenen davon betroffen ist, und die nationale Organisation für seltene Erkrankungen (NORD) schätzt, dass die Zahl näher bei 1 liegt, bei 165.000 bis 200.000 Geburten.
Die meisten Fälle von Apert-Syndrom treten ohne vorherige Familienanamnese auf. Allerdings berichten die NORD auch, dass wenn ein Elternteil die Störung hat, das Kind eine 50-prozentige Chance hat, es zu entwickeln. Diese Statistik gilt für jede Schwangerschaft.
Dieses Syndrom scheint Männer und Frauen gleichermaßen zu betreffen.
Diagnose
Apert-Syndrom kann oft bei der Geburt oder in einem frühen Alter diagnostiziert werden.
Um eine Person mit Apert-Syndrom formell zu diagnostizieren, wird ein Arzt nach den charakteristischen Knochenanomalien suchen, die den Kopf, das Gesicht, die Hände und die Füße betreffen.
Ein Arzt kann eine Schädel-Röntgenaufnahme oder einen CT-Scan des Kopfes durchführen, um die Art der Knochenanomalien zu bestimmen. Molekulargenetische Tests können auch bei der Diagnose helfen.
Behandlung

Die Behandlung des Apert-Syndroms variiert zwischen einzelnen Personen. Ein Arzt wird oft eng mit der Person und ihrer Familie zusammenarbeiten, um einen Plan zur Behandlung ihrer Symptome zu entwickeln.
In einigen Fällen kann ein Arzt eine Operation empfehlen, um den Druck auf das Gehirn der Person zu reduzieren.
Andere chirurgische Optionen sind ebenfalls möglich, beispielsweise um Gesichtsmerkmale neu zu formen oder um verschmolzene Finger oder Zehen zu trennen.
Ein Kind mit Apert-Syndrom wird lebenslange Beobachtungen und Untersuchungen benötigen. Ein Arzt wird auf zusätzliche Komplikationen achten, die durch das Apert-Syndrom verursacht werden, und geeignete Behandlungen vorschlagen.
Häufige Behandlungen für Apert-Syndrom-bedingte Komplikationen sind:
- Sehprobleme beheben
- Therapien zur Bewältigung von Entwicklungs- und Wachstumsverzögerungen
- Zahnbehandlungen, um überfüllte Zähne zu korrigieren
Ausblick
Eine frühzeitige Diagnose bei Babys verbessert die Chancen, einige der Symptome und Komplikationen des Apert-Syndroms erfolgreich zu behandeln.
Das Apert-Syndrom beeinflusst oft nicht die Lebenserwartung einer Person, obwohl Herzprobleme oder andere damit verbundene Zustände zu Komplikationen führen können.
Die langfristigen Aussichten für Menschen mit Apert-Syndrom verbessert sich zusammen mit Fortschritten in der modernen Medizin.
Die Behandlung kann oft die Gesamtaussichten für eine Person mit Apert-Syndrom verbessern.
Ein Kind mit Apert-Syndrom sollte fortlaufend klinisch betreut werden. Dies kann eine Therapie einschließen, die einem Kind und seiner Familie hilft, die täglichen Herausforderungen dieses Syndroms zu bewältigen.