Das DiGeorge-Syndrom, das ansonsten als 22q11.2-Deletionssyndrom bezeichnet wird, ist eine Chromosomenstörung, die typischerweise das 22. Chromosom am q11.2-Standort betrifft, wobei 90% der Fälle dieses Deletionsmerkmal aufweisen.1-3
Dieser Zustand wurde auch als velokardiofaziales Syndrom, konotrunales Syndrom, Shprintzen-Syndrom und CATCH22 sowie viele andere bekannt
Experten schätzen, dass das DiGeorge-Syndrom 1 von 4.000 Menschen betrifft. Aufgrund der Variabilität bei der Präsentation von Symptomen können jedoch viele Menschen unterdiagnostiziert oder falsch diagnostiziert werden.2,3
Das DiGeorge-Syndrom führt dazu, dass sich mehrere Körpersysteme schlecht entwickeln und zu so unterschiedlichen medizinischen Problemen wie Herzfehlern, Verhaltensstörungen und einer Gaumenspalte führen können.
Schnelle Fakten zum DiGeorge-Syndrom
Hier sind einige wichtige Punkte zum DiGeorge-Syndrom. Weitere Details und unterstützende Informationen finden Sie im Hauptartikel.
- DiGeorge-Syndrom ist auch bekannt als Cayler Kardiofazialen Syndrom und Sedlackova-Syndrom
- Der Zustand wird typischerweise als 22q11.2-Deletionssyndrom bezeichnet, da dies am genauesten seine Ursprünge widerspiegelt
- Die Deletion von Genen aus dem 22. Chromosom erfolgt normalerweise zufällig; Die Bedingung wird selten vererbt
- Die Symptome des DiGeorge-Syndroms hängen davon ab, welches Organsystem von der Störung betroffen ist
- DiGeorge-Syndrom wird oft mit einem spezifischen Bluttest diagnostiziert
- Die Behandlung von DiGeorge-Syndrom erfordert in der Regel eine Reihe von Spezialisten aus verschiedenen Bereichen der Medizin.
Ursachen des DiGeorge-Syndroms
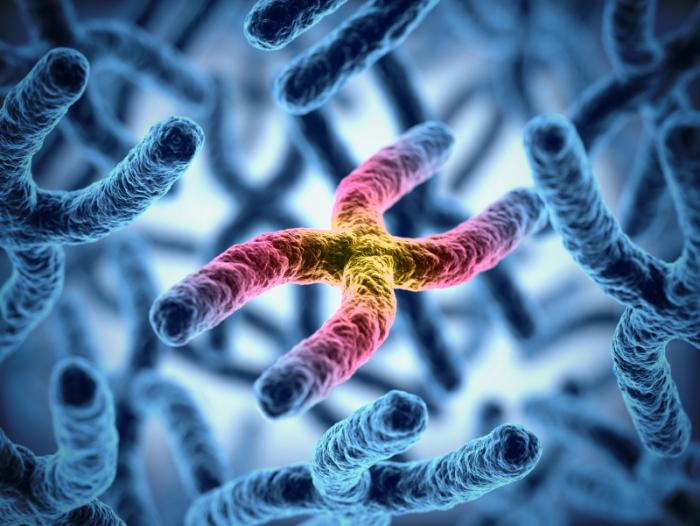
Das DiGeorge-Syndrom wird durch die Deletion des 22q11.2-Segments in einer der beiden Kopien von Chromosom 22 verursacht, was ungefähr 30-40 Gene betrifft. Viele dieser Gene müssen von Wissenschaftlern noch vollständig verstanden werden.1,3
Die Entwicklung des DiGeorge-Syndroms ist typischerweise ein zufälliges Ereignis, das während der Befruchtung auf der mütterlichen oder väterlichen Seite oder während der Zeit der fetalen Entwicklung auftritt.1-3
Die meisten Fälle von DiGeorge-Syndrom sind nicht in der Natur vererbt, da die meisten Menschen mit DiGeorge-Syndrom keine Familiengeschichte der Erkrankung haben, obwohl sie von einem betroffenen Elternteil an ein Kind weitergegeben werden können; Dies tritt bei etwa 10% der DiGeorge-Syndrom-Fälle auf.1,3
Symptome des DiGeorge-Syndroms
Anzeichen und Symptome des DiGeorge-Syndroms sind vielfältig und umfassen: 1-3
- Atembeschwerden
- Mund-, Arm-, Hals- oder Handkrämpfe
- Häufige Infektionen
- Abnormale Gesichtszüge einschließlich unterentwickeltem Kinn, tiefsitzenden Ohren und weit geöffneten Augen
- Gaumenspalte oder andere Gaumenstörungen
- Verzögertes Wachstum
- Schlechter Muskeltonus
- Verzögerte Entwicklung, verzögerte Sprache und andere Lernverzögerungen
- Verhaltensprobleme wie ADHS
- Psychiatrische Erkrankungen wie Schizophrenie
- Autoimmunerkrankungen wie idiopathische Thrombozytopenie Purpura, autoimmune hämolytische Anämie, Autoimmunarthritis und Autoimmunerkrankungen der Schilddrüse
- Parathyroid Drüse Anomalien wie Krampfanfälle, Hypoparathyreoidismus und abnorme Calcium-und Phosphor-Stoffwechsel
- Thymusdrüsenanomalien wie T-Lymphozyten-Defizite
- Blaue Hautfarbe (Zyanose), die sekundär zu schlechter Durchblutung aufgrund von Herzfehlern ist – diese betreffen typischerweise die Aorta und können einen Ventrikelseptumdefekt (Loch zwischen den unteren Herzkammern, Truncus arteriosus (fehlendes Herzgefäß) und Fallot – Tetralogie) einschließen vier abnormale Herzstrukturen).
Andere Symptome können eine Schwerhörigkeit, Sehstörungen und eine veränderte Nierenfunktion sein
Aufgrund der signifikanten Variabilität des DiGeorge-Syndroms werden Typ und Schweregrad der Symptome typischerweise durch das betroffene Organsystem bestimmt.
Das DiGeorge-Syndrom kann bei der Geburt, in der Kindheit oder in der frühen Kindheit auftreten.1
Diagnose des DiGeorge-Syndroms
Das DiGeorge-Syndrom wird am häufigsten mit einem Bluttest diagnostiziert, der als FISH-Analyse bezeichnet wird (Fluorescent In Situ Hybridization) .1,2
Ein Gesundheitsdienstleister wird wahrscheinlich eine FISH-Analyse anfordern, wenn ein Kind Symptome zeigt, die auf das DiGeorge-Syndrom hindeuten, oder wenn ein Kind einen Herzfehler hat – bestimmte Arten von Herzfehlern sind stark mit der Erkrankung verbunden.
Behandlung für DiGeorge-Syndrom
Die Behandlung der Erkrankung hängt von den beteiligten Organsystemen ab und erfordert möglicherweise eine Koordinierung der Versorgung mit vielen Mitgliedern der Gesundheitsversorgung.1,2

Die Koordination der Betreuung durch ein Kind kann eine beliebige Anzahl der folgenden medizinischen Fachkräfte umfassen: 1
- Kinderärzte
- Kardiologen
- Genetiker
- Immunologen
- Spezialisten für Infektionskrankheiten
- Endokrinologen
- Chirurgen
- Therapeuten (beruflich, körperlich, sprachlich, entwicklungsbezogen und geistig).
Verschiedene Bedingungen, die durch das DiGeorge-Syndrom verursacht werden, erfordern unterschiedliche Behandlungsansätze, einschließlich der folgenden: 1,2
- Hypoparathyreoidismus: Behandlungen zur Behandlung von Hypoparathyreoidismus können eine Supplementierung mit Vitamin D oder Kalzium sowie eine Supplementierung mit Parathyroidhormon umfassen
-
- Eine eingeschränkte Funktion der Thymusdrüse kann die Fähigkeit eines Individuums mit DiGeorge-Syndrom zur Bekämpfung einer Infektion beeinträchtigen, und häufige Infektionen können häufig auftreten, gleichgültig ob sie milder oder moderater Natur sind. Die Behandlung und Impfplanung für diese Kinder ist typischerweise die gleiche wie für ihre nicht betroffenen Altersgenossen, und ihre Immunfunktion wird sich typischerweise verbessern, wenn sie älter werden.
- Eine schwere Thymusdysfunktion führt bei einem Kind mit DiGeorge-Syndrom zu einem Risiko für schwere Infektionen, und sie müssen sich einer Thymusgewebetransplantation, einer Knochenmarktransplantation, einer Stammzelltransplantation oder einer Transplantation krankheitskontrollierender Blutzellen unterziehen.
- Thymusdrüsenfunktion:
- Gaumenspalte: chirurgische Reparatur
- Herzfehler: am typischsten mit chirurgischer Reparatur angesprochen
- Entwicklung: Bei der Gesamtentwicklung eines Kindes mit DiGeorge-Syndrom wird eine Vielzahl von Therapeuten befragt (siehe oben)
- Psychische Versorgung: Die Behandlung bestimmter psychischer Störungen kann abhängig von der Diagnose eine Behandlung erfordern.
Prognose des DiGeorge-Syndroms
Derzeit gibt es keine Heilung für DiGeorge-Syndrom. Die Prognose für die Betroffenen mit DiGeorge-Syndrom hängt vom betroffenen Organsystem und der Schwere der Erkrankung ab. Wie bei den meisten medizinischen Bedingungen ist eine frühzeitige Diagnose und Behandlung unerlässlich.1,2
Sprechen Sie mit Ihrem Arzt, wenn Sie der Meinung sind, dass Ihre Schwangerschaft ein Risiko für die Entwicklung des DiGeorge-Syndroms birgt oder wenn Ihr Kind Symptome der Erkrankung zeigt.
Aktuelle Entwicklungen auf dem DiGeorge-Syndrom von MNT News
Auf der Suche nach genetischen Verbindungen zwischen Autismus und anderen Erkrankungen
Die in dieser Woche veröffentlichte Studie zeigt eine starke Beziehung zwischen einem spezifischen Gen, Autismus und dem 22q11.2-Deletionssyndrom. Die Erkenntnisse eröffnen neue Wege der Forschung.